総論
CAHはステロイド合成経路上の酵素欠損により、コルチゾール合成が障害される常染色体劣性遺伝疾患の総称。コルチゾール低下によるネガティブフィードバック解除でACTHが上昇し、副腎皮質が過形成となる。欠損酵素の種類により上流ホルモンの蓄積と下流ホルモンの欠乏が生じる。
21-水酸化酵素欠損症の病態
21-水酸化酵素はプロゲステロン→11-デオキシコルチコステロン(DOC)、17-OHプロゲステロン→11-デオキシコルチゾールへの変換を触媒する。欠損すると:
- 上流に蓄積 — 17-OHプロゲステロン(17-OHP)↑↑ → アンドロゲン経路へ流入 → DHEA・アンドロステンジオン・テストステロン↑
- 下流が欠乏 — コルチゾール↓ → ACTH↑(フィードバック解除) → 副腎過形成
- アルドステロン欠乏(塩類喪失型) — ミネラルコルチコイド経路も障害 → Na喪失・K貯留
病型分類
| 病型 | 酵素活性 | 特徴 | 発症時期 |
|---|---|---|---|
| 塩類喪失型(SW型) | ほぼ消失 | 低Na・高K・脱水クリーゼ+男性化 | 新生児期 |
| 単純男性化型(SV型) | 1〜2%残存 | 男性化のみ、電解質異常なし | 乳幼児〜学童 |
| 非古典型(NC型) | 20〜50%残存 | 軽度男性化、思春期早発・不妊 | 思春期〜成人 |
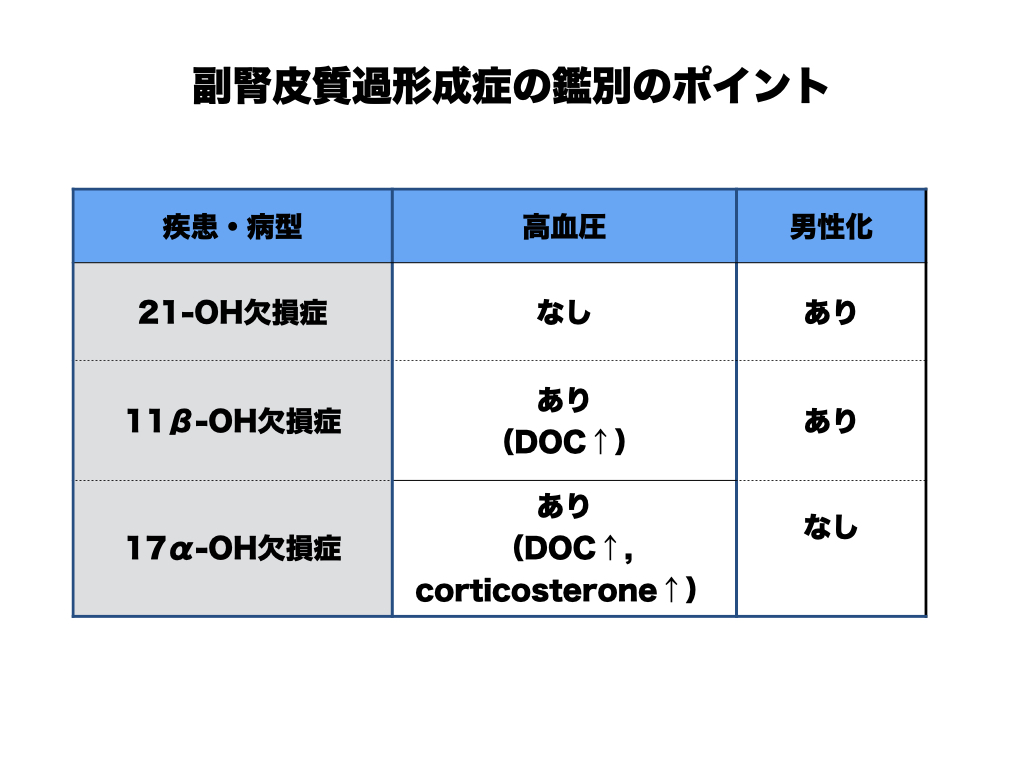
鑑別のポイント
21-水酸化酵素以外の酵素欠損も国試・専門医試験で問われる。高血圧の有無と男性化の有無の組み合わせで主要3病型を鑑別できる。鍵は11-デオキシコルチコステロン(DOC)の蓄積(鉱質コルチコイド作用 → 高血圧)と、アンドロゲン経路への流入の有無(男性化)である。
| 疾患・病型 | 高血圧 | 男性化 |
|---|---|---|
| 21-OH欠損症 | なし | あり |
| 11β-OH欠損症 | あり (DOC↑) | あり |
| 17α-OH欠損症 | あり (DOC↑、corticosterone↑) | なし |
臨床像
- 女性新生児の外性器男性化 — アンドロゲン過剰により陰核肥大・陰唇癒合。性分化疾患(DSD)として出生直後に発見されることも。
- 男性新生児 — 外性器は正常のため見落とされやすく、塩類喪失型クリーゼで発見されることがある。
- 塩類喪失型クリーゼ — 生後2〜4週に嘔吐・体重減少・低Na・高K・脱水・ショック。
- 早発思春期・最終身長低下 — アンドロゲン過剰により骨端線が早期閉鎖。
- 副腎偶発腫瘍(成人例) — 治療不十分な長期例でACTH刺激による副腎過形成結節が発見されることがある。
診断
生後4〜6日のカカト血(ろ紙血)で17-OHPを測定。閾値以上で要精査となる。本邦では1989年より開始。
血中17-OHP高値(基礎値または ACTH 刺激後)が決め手。副腎アンドロゲン(DHEA-S・アンドロステンジオン)も上昇。コルチゾール低値、ACTH高値を伴う。遺伝子解析(CYP21A2変異)で確定。
治療
- グルココルチコイド補充 — ヒドロコルチゾン(小児)またはプレドニゾロン(成人)。ACTH抑制によりアンドロゲン過剰を抑制することが治療の主眼。
- 鉱質コルチコイド補充(塩類喪失型) — フルドロコルチゾン + 食塩補充(乳児期)。
- 急性クリーゼ対応 — 生食静注 + ヒドロコルチゾン100 mg/m² iv。副腎不全クリーゼと同様の対応。
- 外科的治療 — 女性化外性器の形成術(陰核形成・膣形成)。時期・術式は専門施設で慎重に判断。
- モニタリング — 17-OHP・アンドロステンジオン・身長・骨年齢を定期フォロー。過剰治療(クッシング様)にも注意。
過去問
先天性副腎皮質過形成症の維持療法中に発熱を呈した場合、初期対応として適切なのはどれか。
- a水分制限
- b抗菌薬投与
- c利尿薬静注
- d糖質コルチコイドの増量
- eグルコース・インスリン療法
正解を見る · Reveal answer
正解:d
18 歳の女子。無月経を訴えて来院した。二次性徴の遅れがあり、陰毛が欠如。血圧 160/110 mmHg。血清生化学所見:尿素窒素 10 mg/dL、クレアチニン 0.8 mg/dL、Na 148 mEq/L、K 3.0 mEq/L、血漿レニン活性 0.3 ng/mL/h(基準 1.2〜2.5)。
この患者で高値が予想される血中ホルモンをすべて選べ。
- aACTH
- bデヒドロエピアンドロステロン(DHEA)
- cコルチゾール
- dデオキシコルチコステロン(DOC)
- eプロゲステロン
正解を見る · Reveal answer
正解:a・d・e